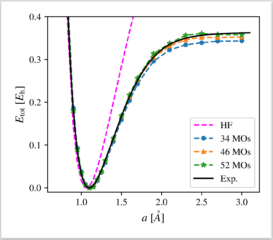
We apply a recently proposed computational protocol for a neural-network-supported configuration interaction calculation to the paradigmatic N2 molecule. By comparison of correlation energy, binding energy, and the full dissociation curve to experimental and full CI benchmarks, we demonstrate the applicability and robustness of our approach for the first time in the context of molecular systems, and offer thereby a new complementary tool in the family of machine-learning-based computation methods. The main advantage of the method lies in the efficiency of the neural-network-selected many-body basis set. Specifically, we approximate full CI results obtained on bases of ≈1010Slater Determinants with only ≈105 determinants with good accuracy. The high efficiency of the NN CI approach underlines its potential for broader applications such as structural optimizations and even computation of spectroscopic observables in systems for which computational resources are a limiting factor.
Preprint: Revisiting N2 with Neural-Network-Supported CI